键德测试测量|挑选国产探针台厂家要考虑什么?
探针台是电子研发和生产中不可或缺的测试工具,主要用于测试和分析电子产品。过去,国外品牌的探针台在市场上占据主导地位,但近...
科技
3 小时前

写在前面
这个包最优雅的地方在于交互式,所以学习的主要目的也就是交互式的实践。交互图可以很好的探索数据,但一般不支持输出矢量图,不方便下游编辑和修改和用于发表。如果你找到了导出矢量图方法,请留言。
首先我查看了作者的介绍:这是参与发表过Nature的优秀青年,处于事业的快速上升期。文献详见:https://scholar.google.com/citations?user=pLIF9tUAAAAJ&hl=zh-CN
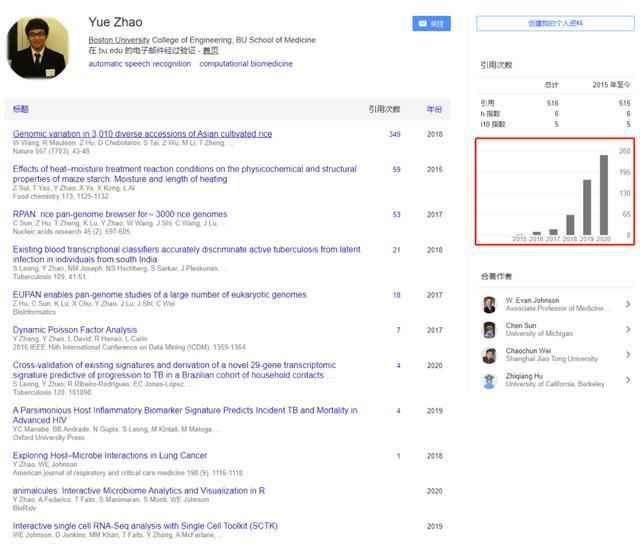
作者信息
Yue Zhao 1,2
Anthony Federico 1,2
Evan Johnson 1,2
1Boston University School of Medicine, Boston, MA
2Bioinformatics Program, Boston University, Boston, MA
介绍
animalcules包专注于微生物组数据的统一框架,然后包装目前比较流行的分析方法和手段,作者说传统的alpha和beta多样性已经太泛滥了,于是将生物标记物分析的方法引入*animalcules*包中,然后使用交互式的图表来分析和了解数据,更为强大。大家一定要注意,这类框架都使用的S4类对象,不然不会如此方便。
以下教程主要参考软件帮助手册,原文详见:https://bioconductor.org/packages/release/bioc/vignettes/animalcules/inst/doc/animalcules.html
R包安装
软件依赖关系众多,如有些自动安装失败的包,还需要在RStudio中Packages管理中心手动安装。
#--安装BiocManager用于安装Bioconductor的R包
if (!requireNamespace("BiocManager", quietly=TRUE))
install.packages("BiocManager")
#--BiocManager安装Bioconductor中的animalcules包
if (!requireNamespace("animalcules", quietly=TRUE))
BiocManager::install("compbiomed/animalcules")
## 安装github版本
if (!requireNamespace("devtools", quietly=TRUE))
install.packages("devtools")
if (!requireNamespace("animalcules", quietly=TRUE))
devtools::install_github("compbiomed/animalcules")
#下面演示构造MultiAssayExperiment对象时,用到了里面内置的phyloseq对象数据集
if (!requireNamespace("ggClusterNet", quietly=TRUE))
devtools::install_github("taowenmicro/ggClusterNet")
#--载入R包
library(animalcules)
library(SummarizedExperiment)
library(MultiAssayExperiment)
data_dir = system.file("extdata/MAE.rds", package = "animalcules")
MAE = readRDS(data_dir)
关于数据格式的必要了解
总之一句话,想要更方便,就要多封装,一致的格式,统一的格式。
作者默认数据解析MAE:其实核心就是SummarizedExperiment数据,多个SummarizedExperiment就是MultiAssayExperiment。这可是多组学数据整合的一个比较好的框架呀。下面我们简单介绍一下
多组学数据处理思路
组学实验越来越普遍、为实验设计,数据整合和分析增加了复杂性。R和Bioconductor为统计分析和可视化提供了一个通用框架,为各种高通量数据类型提供了专门的数据类,但缺乏对多组学实验进行整合分析的方法。
MultiAssayExperiment
多组学实验的整合R包,MultiAssayExperiment,为多种多样的基因组数据提供一致的表示,存储和操作。
MultiAssayExperiment(https://bioconductor.org/packages/MultiAssayExperiment)引入了一个面向对象的S4类对象,定义了用于表示多组学实验的通用数据结构。它有三个关键组成部分:
(i)colData,一个包含患者或细胞系水平的特征(如病理学和组织学)的“主要”数据集;
(ii)ExperimentList,主要数据存储对象,可以包括多组的数据。
(iii)sampleMap,用于全部数据之间联系起来的map文件。
(1)构造函数和相关的有效性检查,简化创建MultiAssayExperiment对象,同时允许灵活地表示复杂的实验。
(2)允许进行数据选择的子集操作。
MultiAssayExperiment核心数据基于SummarizedExperiment,同时支持异质性的多组学数据。MultiAssayExperiment类和方法提供了一个灵活的框架,用于整合和分析重叠样本的互补分析。它集成了任何支持基本子集和维度名称的数据类,因此默认情况下支持许多数据类,而不需要额外的调整。
#-关于A MultiAssayExperiment object对象的一些基础操作
#assays部分
# assays(MAE)# 提取数据矩阵部分文件,这是一个list,所以提取每个矩阵需要继续
#--下面提取第一个矩阵,这有什么用呢,类似多组学数据,用于操作更加简便。
# assays(MAE)[[1]]# 第二个对象类似
# assays(MAE)[[2]]
#--colData,也就是对数据矩阵列名的注释信息,类似于phyloseq对象中的map文件
colData(MAE)
#--查看子对象数量,都是s4类对象,可以单独提取
#--就单个的S4类对象进行各部分数据的提取
microbe otu_table tax_table map
构造MultiAssayExperiment对象
使用phyloseq对象构建MultiAssayExperiment对象
至于如何构造phyloseq文件,可以点击查看。想要具体学习什么是phyloseq对象,可以点击
# 如何构建这个MAE对象呢?
#--得到phyloserq对象并提取必要数据信息
library(ggClusterNet)
library(phyloseq)
ps
otu = as.data.frame(t(vegan_otu(ps)))
head(otu)
tax = as.data.frame((vegan_tax(ps)))
head(tax)
map = sample_data(ps)
head(map)
#--首先构造SummarizedExperiment对象,比较简单,类似phyloseq对象
micro colData=map,
rowData=tax)
# 将SummarizedExperiment对象封装成为ExperimentList
mlist mlist[[1]] = micro
names(mlist) = "MicrobeGenetics"# 注意必须命名,否则无法区分每个部分数据组
# 构造不同数据组之间的记录文件
gistmap primary = row.names(map),
colname = row.names(map),
stringsAsFactors = FALSE)
maplistowe sampMapowe
# colData文件为分组文件,数据框即可,本案例只有一个微生物组数据,所以直接用map文件就可以了。
#-下面就直接构建了MultiAssayExperiment文件
mae sampleMap = sampMapowe)
运行Shiny app
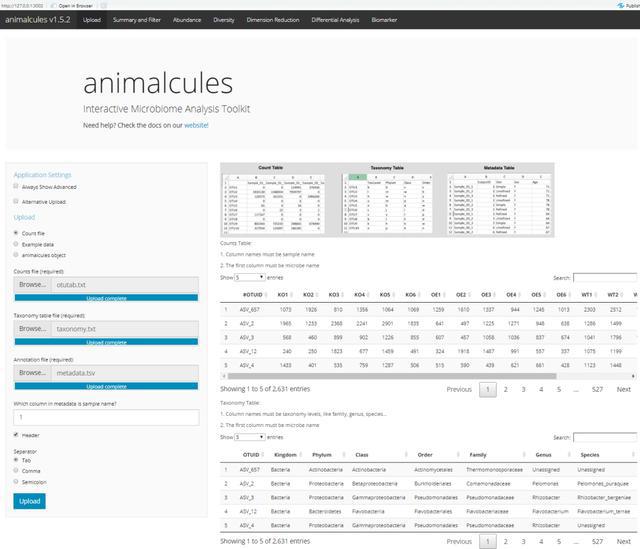
这里我们准备了自己的示例文件,也是为保证尽可能的将这个工具变成一种普通工具,供大家使用。
run_animalcules### 基础统计
往下的部分,我会同时运行Shiny和官方代码教程,并作比对,但是不会深入挖掘,仅仅会帮助大家完成官方教程和提出一些建议。其次虽然我们也构建MAE文件,但是由于作者代码有一些小瑕疵,所以使用官方数据做演示。
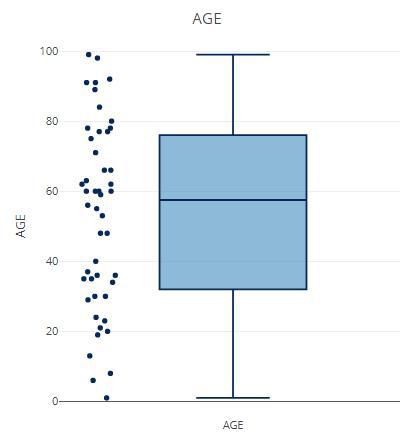
这部分用于统计每个样本中OTU的数量,并做两种方式可视化:频率曲线,箱线+散点图;如果使用shiny程序的话,直接可以展示表格。

此外,可以按照微生物分类水平合并OTU数据:

- samples_discard : 需要去除样本的id
```{R}
# ?filter_summary_pie_box
p samples_discard = c("subject_2", "subject_4"),
filter_type = "By Metadata",
sample_condition = "AGE")
p
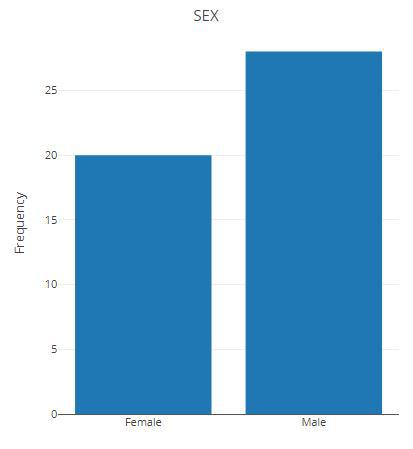
更换分组,重新统计。
p samples_discard = c("subject_2", "subject_4"),
filter_type = "By Metadata",
sample_condition = "SEX")
p
#--提取子集,并且提取map文件
microbe samples result sample_condition="AGE",
new_label="AGE_GROUP",
bin_breaks=c(0,55,75,100),
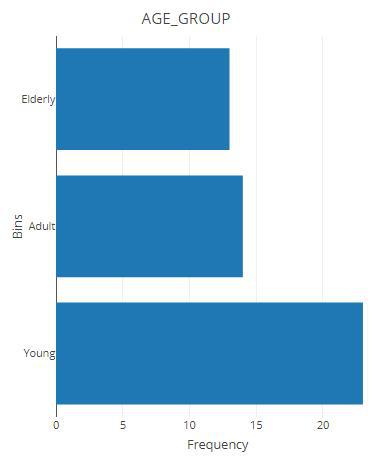
bin_labels=c('Young','Adult',"Elderly"))
head(result$sam_table)result$plot.binned
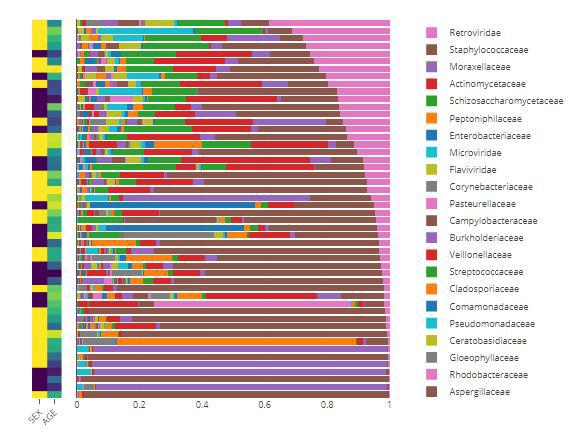
丰度展示-堆叠柱状图
通过tax_level选择某个分类等级,通过 sample_conditions 选择需要添加的分组标签。值得注意的是这里可以对堆叠柱状图排序的,通过order_organisms来指定,默认丰度从高到低。这里从源代码来看就是通过改变factor来实现的,所以图例第一个也是排序的这个微生物。
p tax_level="family",
order_organisms=c('Retroviridae'),
sort_by="organisms",
sample_conditions=c('SEX', 'AGE'),
show_legend=TRUE)
p
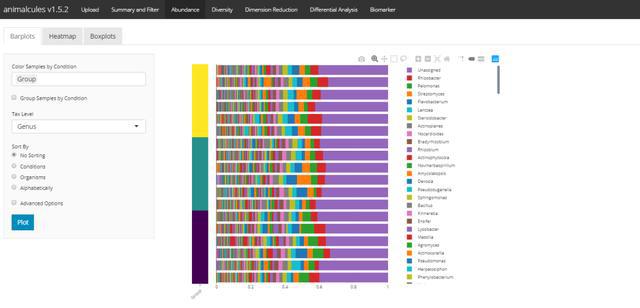
shiny版本:
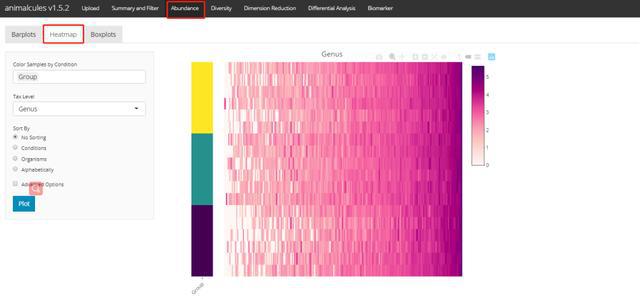
丰度-热图p tax_level="genus",
sort_by="conditions",
sample_conditions=c("SEX", "AGE"))
p
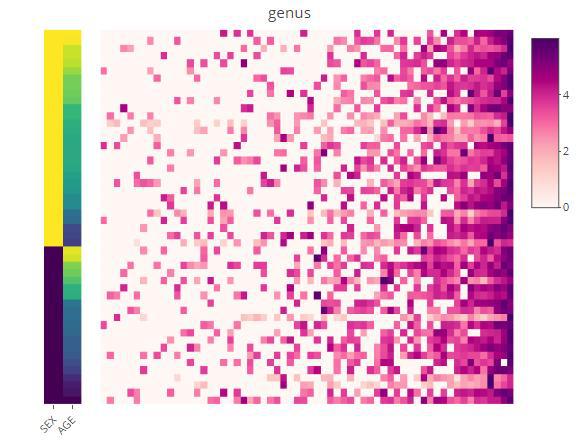
shiny版本:
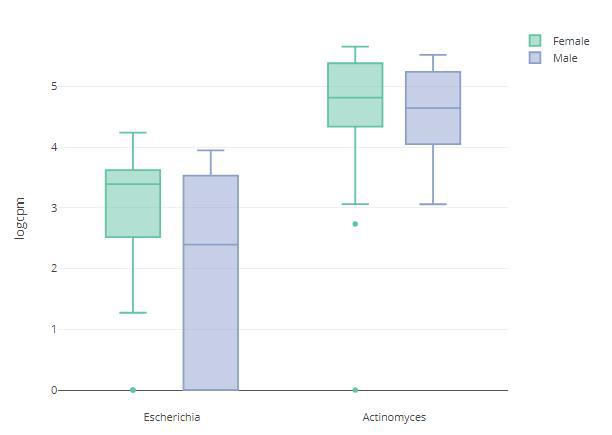
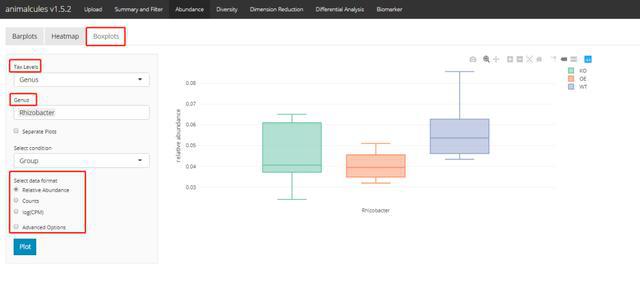
丰度-箱线图p tax_level="genus",
organisms=c("Escherichia", "Actinomyces"),
condition="SEX",
datatype="logcpm")
p
shiny版本:
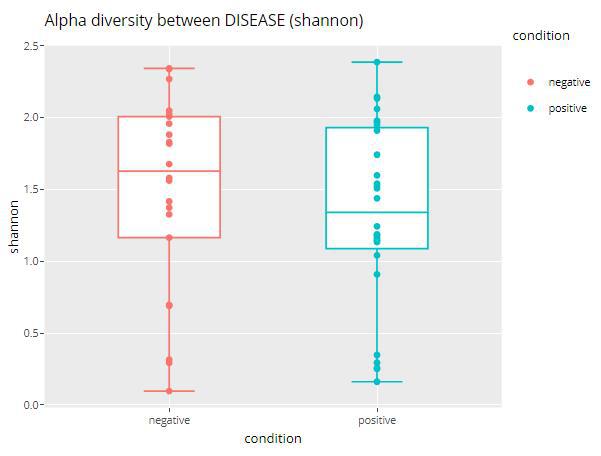
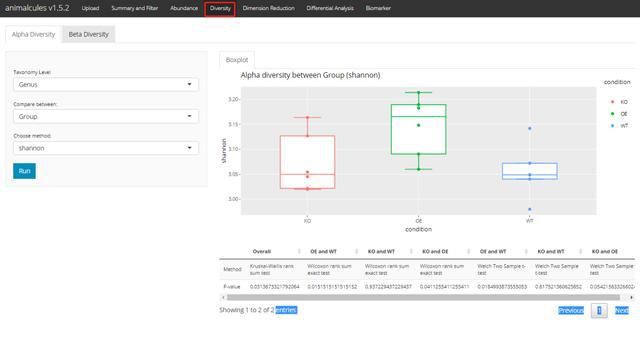
Alpha多样性
这里只有四个多样性指标。然后通过箱线+散点图展示。
alpha_div_boxplot(MAE = MAE,
tax_level = "genus",
condition = "DISEASE",
alpha_metric = "shannon")
对多样性进行统计检验。这里可选的是”Wilcoxon rank sum test”, “T-test”, “Kruskal-Wallis”这三种方法。
# ?do_alpha_div_test
do_alpha_div_test(MAE = MAE,
tax_level = "genus",
condition = "DISEASE",
alpha_metric = "shannon",
alpha_stat = "T-test")
shiny版本:
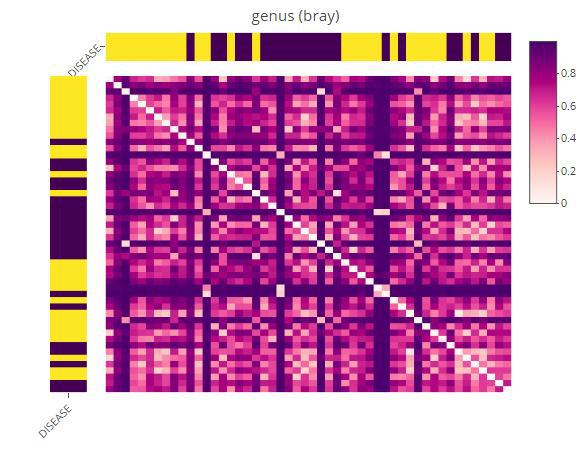
Beta多样性-聚类距离热图diversity_beta_heatmap(MAE = MAE,
tax_level = 'genus',
input_beta_method = "bray",
input_bdhm_select_conditions = 'DISEASE',
input_bdhm_sort_by = 'condition')
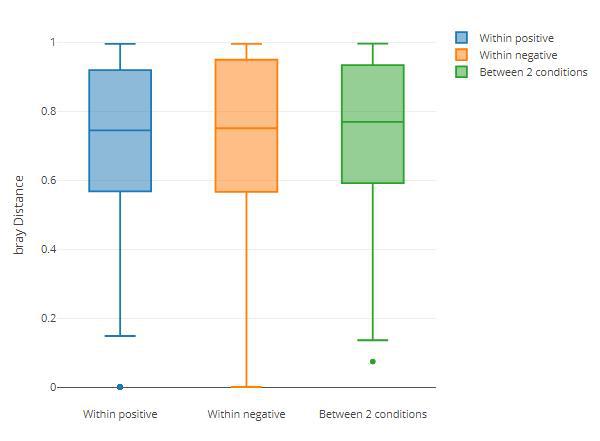
Beta多样性-组间距离箱线图
其次通过组内距离和组间距离的箱线图展示
diversity_beta_boxplot(MAE = MAE,
tax_level = 'genus',
input_beta_method = "bray",
input_select_beta_condition = 'DISEASE')
再有就是统计检验,共有三种方法可以选择:PERMANOVA,Kruskal-Wallis,Wilcoxon test。但是只有两种距离可供选择,其次就是两两比较不能实现。
# ?diversity_beta_test
diversity_beta_test(MAE = MAE,
tax_level = 'genus',
input_beta_method = "bray",
input_select_beta_condition = 'DISEASE',
input_select_beta_stat_method = 'PERMANOVA',
input_num_permutation_permanova = 999)
shiny版本:
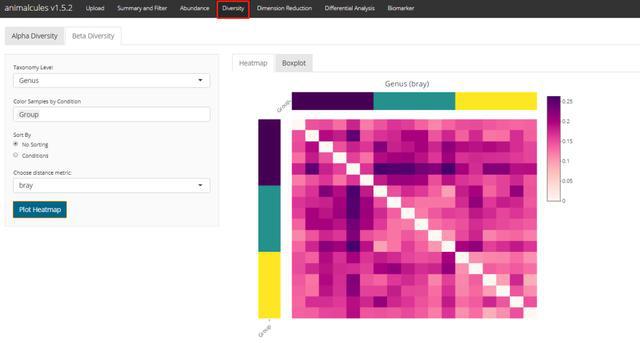
shiny版本(error):
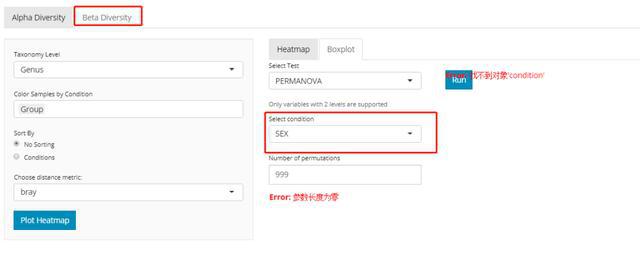
排序PCAresult tax_level="genus",
color="AGE",
shape="DISEASE",
pcx=1,
pcy=2,
datatype="logcpm")
result$plot
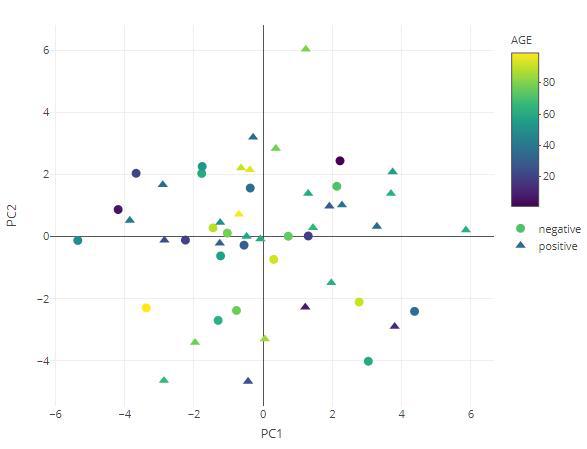
PCoAresult tax_level="genus",
color="AGE",
shape="DISEASE",
axx=1,
axy=2,
method="bray")
result$plot
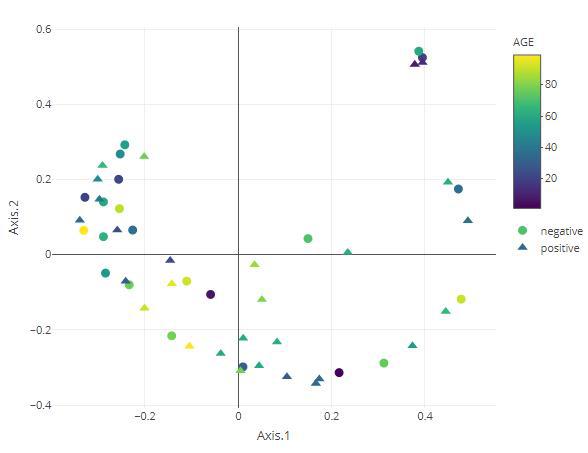
UMAPresult tax_level="genus",
color="AGE",
shape="DISEASE",
cx=1,
cy=2,
n_neighbors=15,
metric="euclidean",
datatype="logcpm")
result$plot
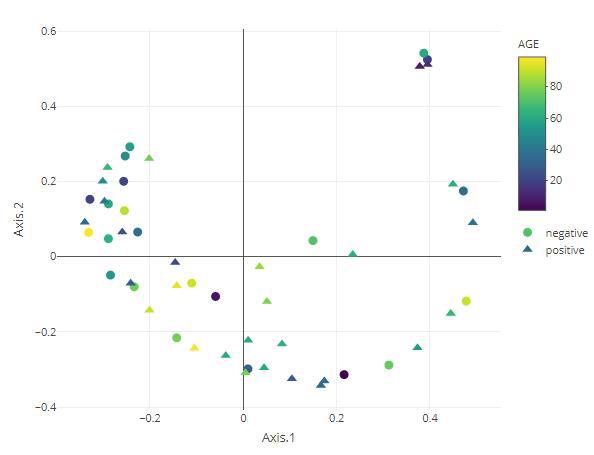
t-SNE
除了二维图形展示还可以进行三维图形的展示。
result tax_level="phylum",
color="AGE",
shape="GROUP",
k="3D",
initial_dims=30,
perplexity=10,
datatype="logcpm")
result$plot
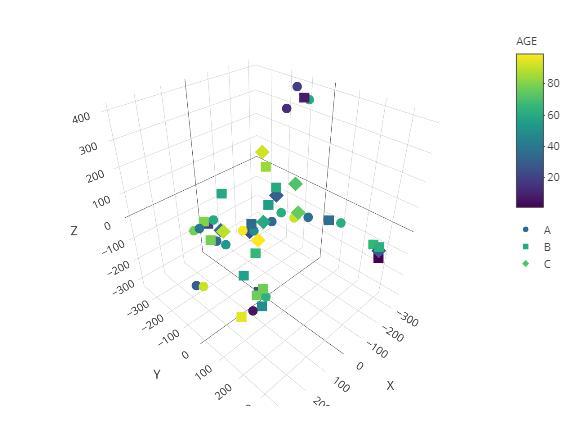
shiny版本:
差异分析p tax_level="phylum",
input_da_condition=c("DISEASE"),
min_num_filter = 2,
input_da_padj_cutoff = 0.5)
p
shiny版本:
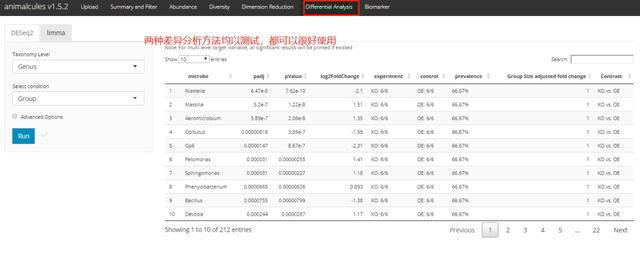
生物标记物分析
这里可选的方法有两个:”logistic regression”, “random forest”。
# ?find_biomarker
p tax_level = "genus",
input_select_target_biomarker = c("SEX"),
nfolds = 3,
nrepeats = 3,
seed = 99,
percent_top_biomarker = 0.2,
model_name = "logistic regression")
p$biomarker
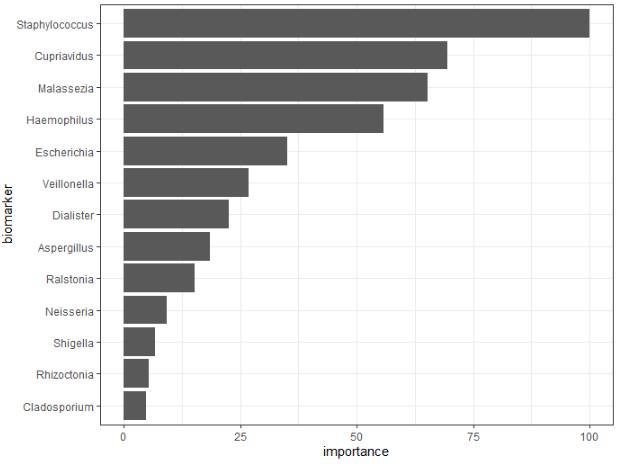
对重要变量可视化。
# importance plot
p$importance_plot
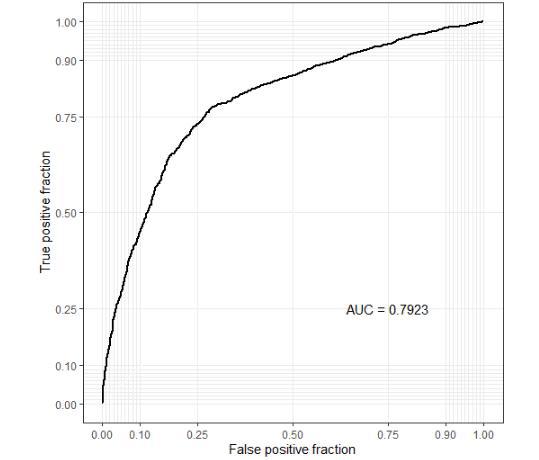
ROC曲线准确度评估。注意ROC曲线只能对二分变量进行操作。
p$roc_plot
shiny版本(error):
运行环境sessionInfoR version 4.1.0 (2021-05-18)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 19042)
Matrix products: default
locale:
[1] LC_COLLATE=Chinese (Simplified)_China.936
[2] LC_CTYPE=Chinese (Simplified)_China.936
[3] LC_MONETARY=Chinese (Simplified)_China.936
[4] LC_NUMERIC=C
[5] LC_TIME=Chinese (Simplified)_China.936
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] caret_6.0-88 biomformat_1.20.0
[3] magrittr_2.0.1 dplyr_1.0.6
[5] vegan_2.5-7 lattice_0.20-44
[7] permute_0.9-5 plotly_4.9.3
[9] ggplot2_3.3.3 shinyjs_2.0.0
[11] shiny_1.6.0 phyloseq_1.36.0
[13] ggClusterNet_0.1.0 MultiAssayExperiment_1.18.0
[15] SummarizedExperiment_1.22.0 Biobase_2.52.0
[17] GenomicRanges_1.44.0 GenomeInfoDb_1.28.0
[19] IRanges_2.26.0 S4Vectors_0.30.0
[21] BiocGenerics_0.38.0 MatrixGenerics_1.4.0
[23] matrixStats_0.58.0 animalcules_1.9.1
loaded via a namespace (and not attached):
[1] utf8_1.2.1 reticulate_1.20 tidyselect_1.1.1
[4] RSQLite_2.2.7 AnnotationDbi_1.54.0 htmlwidgets_1.5.3
[7] ranger_0.12.1 grid_4.1.0 BiocParallel_1.26.0
[10] covr_3.5.1 pROC_1.17.0.1 devtools_2.4.2
[13] munsell_0.5.0 codetools_0.2-18 DT_0.18
[16] umap_0.2.7.0 rentrez_1.2.3 withr_2.4.2
[19] colorspace_2.0-1 knitr_1.33 rstudioapi_0.13
[22] labeling_0.4.2 GenomeInfoDbData_1.2.6 plotROC_2.2.1
[25] bit64_4.0.5 farver_2.1.0 rhdf5_2.36.0
[28] rprojroot_2.0.2 vctrs_0.3.8 generics_0.1.0
[31] ipred_0.9-11 xfun_0.23 R6_2.5.0
[34] locfit_1.5-9.4 rex_1.2.0 bitops_1.0-7
[37] rhdf5filters_1.4.0 cachem_1.0.5 DelayedArray_0.18.0
[40] assertthat_0.2.1 promises_1.2.0.1 scales_1.1.1
[43] nnet_7.3-16 gtable_0.3.0 processx_3.5.2
[46] timeDate_3043.102 rlang_0.4.11 genefilter_1.74.0
[49] splines_4.1.0 lazyeval_0.2.2 ModelMetrics_1.2.2.2
[52] GUniFrac_1.2 BiocManager_1.30.15 yaml_2.2.1
[55] reshape2_1.4.4 crosstalk_1.1.1 httpuv_1.6.1
[58] tools_4.1.0 lava_1.6.9 usethis_2.0.1
[61] ellipsis_0.3.2 jquerylib_0.1.4 RColorBrewer_1.1-2
[64] proxy_0.4-26 sessioninfo_1.1.1 Rcpp_1.0.6
[67] plyr_1.8.6 progress_1.2.2 zlibbioc_1.38.0
[70] purrr_0.3.4 RCurl_1.98-1.3 ps_1.6.0
[73] prettyunits_1.1.1 rpart_4.1-15 openssl_1.4.4
[76] cluster_2.1.2 fs_1.5.0 data.table_1.14.0
[79] RSpectra_0.16-0 pkgload_1.2.1 hms_1.1.0
[82] mime_0.10 evaluate_0.14 xtable_1.8-4
[85] XML_3.99-0.6 shape_1.4.6 testthat_3.0.2
[88] compiler_4.1.0 tibble_3.1.2 crayon_1.4.1
[91] htmltools_0.5.1.1 mgcv_1.8-35 later_1.2.0
[94] tidyr_1.1.3 geneplotter_1.70.0 lubridate_1.7.10
[97] DBI_1.1.1 MASS_7.3-54 Matrix_1.3-3
[100] ade4_1.7-16 cli_2.5.0 gower_0.2.2
[103] igraph_1.2.6 forcats_0.5.1 pkgconfig_2.0.3
[106] recipes_0.1.16 foreach_1.5.1 annotate_1.70.0
[109] bslib_0.2.5.1 multtest_2.48.0 XVector_0.32.0
[112] prodlim_2019.11.13 stringr_1.4.0 callr_3.7.0
[115] digest_0.6.27 tsne_0.1-3 Biostrings_2.60.0
[118] rmarkdown_2.8 curl_4.3.1 lifecycle_1.0.0
[121] nlme_3.1-152 jsonlite_1.7.2 Rhdf5lib_1.14.0
[124] desc_1.3.0 viridisLite_0.4.0 askpass_1.1
[127] limma_3.48.0 fansi_0.5.0 pillar_1.6.1
[130] KEGGREST_1.32.0 fastmap_1.1.0 httr_1.4.2
[133] pkgbuild_1.2.0 survival_3.2-11 glue_1.4.2
[136] remotes_2.4.0 png_0.1-7 iterators_1.0.13
[139] glmnet_4.1-1 bit_4.0.4 class_7.3-19
[142] stringi_1.6.2 sass_0.4.0 blob_1.2.1
[145] DESeq2_1.32.0 memoise_2.0.0 e1071_1.7-7
[148] ape_5.5
https://bioconductor.org/packages/release/bioc/vignettes/animalcules/inst/doc/animalcules.html
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
来源:我们的百变生活
想查看更多微信群的小伙伴, 可以点击上面导航栏的微信群 或者下面按钮!
点我查看更多微信群
分享

探针台是电子研发和生产中不可或缺的测试工具,主要用于测试和分析电子产品。过去,国外品牌的探针台在市场上占据主导地位,但近...

杭钢集团与国内算力装备头部企业共建的年产10万台算力装备生产线日前投产,全省国企算力装备首个自主品牌“华光宏”亮相。“重...

快科技4月20日消息,据报道,清华大学科研团队在微型机器人领域取得重大突破,成功研制出一种具有"变形金刚"特性的薄膜状微...

快科技4月20日消息,据报道,山西中北测控科技有限公司在工业测控网络领域取得重大突破,其自主研发的SharkNet工业测...

2025年4月20日,当42.195公里的山海赛道与5G-A技术共振,青岛马拉松不止是一场奔跑的盛宴,更成为智慧城市的鲜...
据央视新闻报道,4月19日,全球首次“人机共跑”半程马拉松在北京亦庄开跑。20支人形机器人参赛队和人类选手同时出发,共跑...